
Дорогі читачі! Сьогодні я хочу розповісти вам про переваги банного комплексу «Мартишев». Якщо ви

Приветствую всех любителей маникюра! Сегодня я хочу поделиться с вами удивительной находкой – интернет-магазином,

Кофемашины стали неотъемлемой частью нашей повседневной жизни, обеспечивая нам возможность наслаждаться вкусным кофе прямо

Пансионат Ноев Ковчег для пожилых людей — это специализированное учреждение, которое предоставляет услуги по

У світі все більше підприємців прагнуть розширити свої ділові можливості за межами своєї країни.

Гидроизоляция крыши играет ключевую роль в защите зданий от вредного воздействия воды. Она предотвращает
Накладні натуральні хвости з кріпленням на резинці або стрічці можна використовувати соло, а також
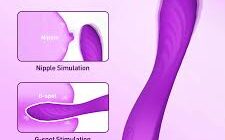
Жінки століттями шукали способи отримати максимальне задоволення від сексу. Однак зі зростаючою популярністю індустрії

Сьогодні силіконові форми стали популярним та невід’ємним елементом у кожній кухні. Вони пропонують широкий

Алюмінієві вікна https://dobroplast.com.ua/ – одне із найпопулярніших рішень при виборі склопакетів. І це невипадково!